DNA-Sequenzierung und die Analyse genetischer Daten sind essenzielle Methoden in den biologischen Wissenschaften und der Gesundheitsversorgung geworden. Heutzutage stehen dafür mehr Sequenzdaten zur Verfügung als jemals zuvor. Dank der Weiterentwicklung der Sequenziertechnologie und sinkender Kosten werden es immer mehr – und das in einem immer rasanteren Tempo.
Große Mengen von Short-Read-Sequenzen werden täglich analysiert, um die Genome der zugrundeliegenden Proben zu rekonstruieren und genetische Variation zu identifizieren. Dabei wird die primäre Analyse, welche im Labor stattfindet und die Probenentnahme, -aufbereitung und -sequenzierung umfasst, von der sekundären Analyse, welche das Sequenzalignment (Alignment) und die Identifikation von genetischen Varianten (Variant Calling) beinhaltet, unterschieden. Diese sekundäre Analyse ist sehr rechenintensiv und wird daher typischerweise nur mit sehr leistungsfähigen Servern auf High Performance Computing (HPC) Infrastruktur durchgeführt. Etablierte Tools für das Alignment und das Variant Calling, welche den de facto Goldstandard darstellen, wurden dabei für eine CPU-basierte Architektur entwickelt. Das sequenzielle Prozessieren auf CPUs ist jedoch zu einem Flaschenhals bei der Analyse von großen Mengen genetischer Daten geworden, sodass sowohl das Alignment als auch das Variant Calling stark von einer effizienten Parallelisierung profitieren. Die Verwendung von GPUs in diesem Kontext ermöglicht eine Steigerung der Performance jenseits von allem, was mit einer CPU-basierten Architektur möglich wäre.
PoC – GPU-beschleunigte Implementierungen
NVIDIA Clara Parabricks ist ein Framework von skalierbaren, GPU-beschleunigten Implementierungen der Standard-Tools zur Analyse genetischer Daten. Im Rahmen eines Proof of Concept (POC) wurde NVIDIA Clara Parabricks auf einem DELL PowerEdge XE8545 in eine Whole-Genome-Germline-Pipeline integriert und mit einer CPU-basierten analogen Pipeline verglichen . Ziel dieses Benchmarks war ein Vergleich der Performance beider Ansätze auf demselben System. Für den POC wurden ausschließlich öffentlich verfügbare Whole-Genome-Datensätze einer sehr ausführlich studierten Zelllinie (NA12878) in unterschiedlicher Größe und Sequenziertiefe (10x, 30x, 50x, 100x und 200x) verwendet. Die Performance der GPU-basierte Pipeline wurde unter Verwendung von einer, zwei, drei und aller vier verfügbaren NVIDIA A100 80GB GPUs getestet. Die CPU-basierte Pipeline wurde indessen unter Verwendung aller zu Verfügung stehen Systemressourcen ausgeführt. Die GPU-basierte Pipeline war bis zu 68 Mal schneller als die CPU -basierte und die Steigerung der Performance war bei Verwendung von mehr GPUs beinahe linear. Die effizienteste Art der Analyse auf dem getesteten Setup war die Verwendung von einer GPU pro Analyse-Lauf und vier parallellaufende Analysen. Der POC demonstriert die große Steigerung der Perfomance für Whole-Genome-Germline-Analysen bei Verwendung eines GPU-optimierten Frameworks wie NVIDIA Clara Parabricks. Damit können auf einem DELL XE8545 mehr als 38 der 30x-Datensätze, mehr als 22 der 50x-Datensätze, mehr als 12 der 100x-Datensätze und mehr als 6 der 200x-Datensätze innerhalb von 24 Stunden analysiert werden.
Verwendete Methoden zur vergleichenden Analyse genetischer Daten
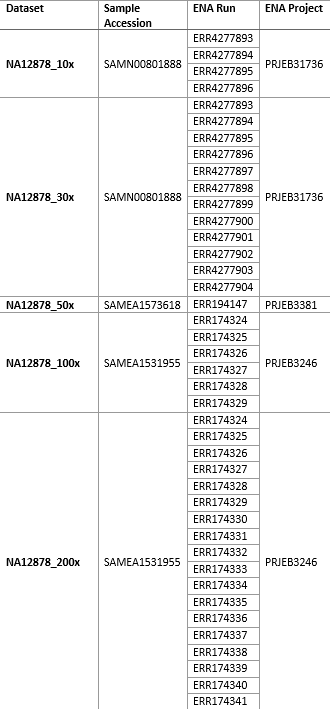
Daten
Verwendet wurden Short-Read-Sequenzdaten (FASTQ) aus zwei publizierten Studien, in denen mehrere Proben der Zelllinie NA12878 sequenziert wurden. Alle Daten wurden über das European Nucleotide Archive (ENA) bezogen.
Die Probe SAMN00801888 wurde für das 1000 Genomes Project Phase 3 mit einer durchschnittlichen Sequenztiefe von 30x über das gesamte Genom sequenziert[1]. Die FASTQ-Dateien aus sechs Sequenzierungsläufen aus dieser Studie sind über das ENA-Projekt PRJEB31736 verfügbar. Um ein 10x-Datensatz herzustellen, wurden die FASTQ-Dateien aus vier Läufen verwendet, für den 30x-Datensatz die FASTQ-Dateien aller Läufe.
Für größere Datensätze wurden Sequenzdaten der Proben SAMEA1573618 und SAMEA1531955 verwendet. Diese wurden in einer Studie zur Erstellung von Referenz-Daten über genetische Variation in ausreichender Tiefe sequenziert[2]. Die FASTQ-Dateien aus einem Sequenzierungslauf, der in dieser Studie mit der Probe SAMEA1573618 durchgeführt wurde, sind über das ENA-Projekt PRJEB3381 verfügbar und wurden für den 50x-Datensatz verwendet. Die Probe SAMEA1531955 wurde in dieser Studie in insgesamt 18 Läufen sequenziert, die Daten sind über das ENA-Projekt PRJEB3246 verfügbar. Die FASTQ-Dateien aus sechs der 18 Läufe wurden zur Erstellung eines 100x-Datensatzes verwendet und die Dateien aus allen 18 Läufen für die Erstellung eines 200x-Datensatzes.
Hardware
Der verwendete DELL PowerEdge XE8545 Server besitzt eine AMD EPYC CPU der dritten Generation und vier NVIDIA A100 Tensor Core GPUs mit jeweils 80GB Grafikspeicher, die über NVLink verbunden sind. Das System verfügt über insgesamt 1 TB Arbeitsspeicher (16x 64GB DDR4 RAM 3200MT) und 7 TB Massenspeicher für Analysen (4x 1788,5 GB PCIe SSD), neben ca 1 TB Massenspeicher für das Betriebssystem (2x 447,13 GB SATA SSD).
Software
Für die Prozessierung der Daten wurde Parabricks Version 3.7.0-1.ampere als Docker-Container verwendet. Die Docker-Container wurden so konfiguriert, dass sie nur die jeweils zu verwendende Anzahl von GPUs zur Verfügung hatten (also je nach Analyse eine, zwei, drei oder alle vier GPUs). Alle anderen Systemressourcen wurden nicht eingeschränkt. Während der Prozessierung der Daten wurden keine anderen Arbeiten auf dem System durchgeführt. Die verwendete Pipeline nutzt die Parabricks-Tools fq2bam und haplotypecaller. Jeder Schritt der Pipeline wurde automatisiert über ein BASH-Skript gestartet. Die CPU-basierte Pipeline verwendet bwa-mem v0.7.15 und GATK v4.2.5.
Ergebnisse
Laufzeiten und Performance
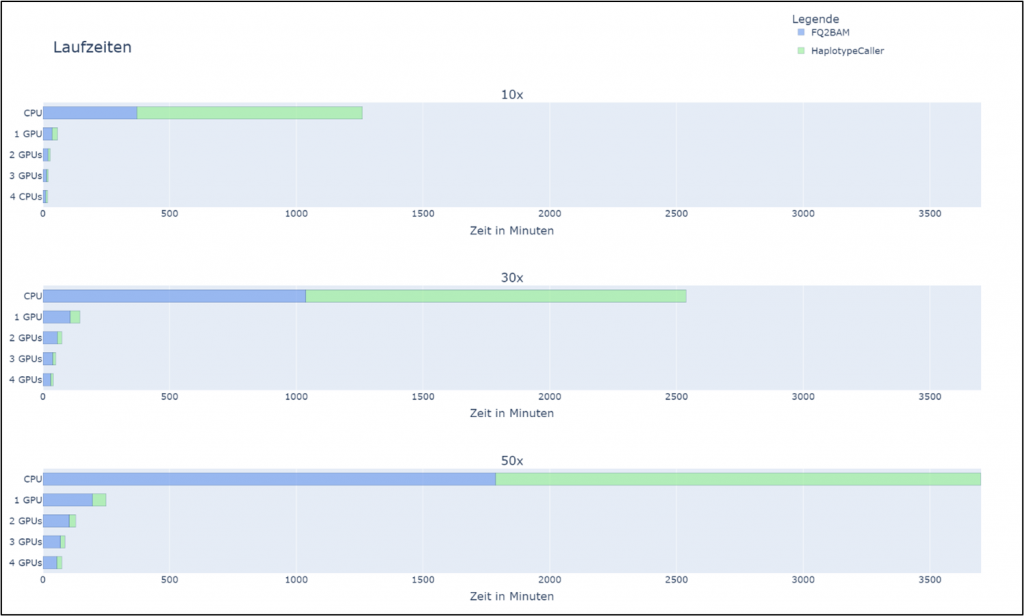
Für einen Vergleich einer Referenz-Performance wurde der 30x-Datensatz verwendet, da es sich dabei um die am häufigsten verwendete Sequenziertiefe eines Datensatzes handelt. Die verwendete CPU-basierte Pipeline, in der bwa-mem direkt von der Kommandozeile gestartet wird, brauchte über 42 Stunden, um den 30x-Datensatz zu prozessieren (Tabelle 1). Die GPU-basierte Pipeline konnte die Laufzeit unter der Verwendung von Parabricks drastisch auf unter 3 Stunden reduzieren.
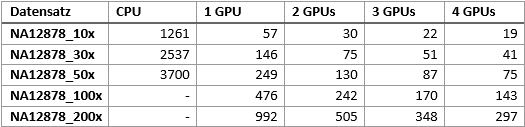
Die Verwendung der Systemressourcen variierte während der Prozessierung (Abbildung 1). Die Auslastung der GPUs war während des ersten Teils der Prozessierung, dem Alignment, bei nahezu 100%, fiel jedoch in den darauffolgenden Schritten drastisch ab. Die Menge des verwendeten GPU-Speichers änderte sich entsprechend der Größe des prozessierten Datensatzes, überschritt jedoch niemals 50% des zu Verfügung stehenden Speichers.
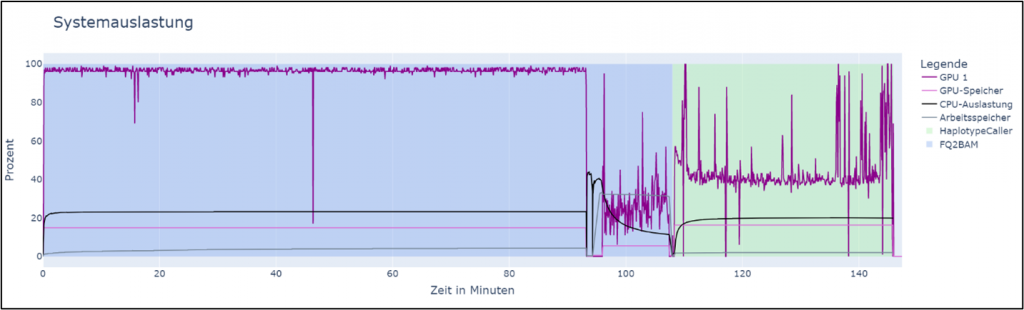
Skalierbarkeit der GPU-Pipeline
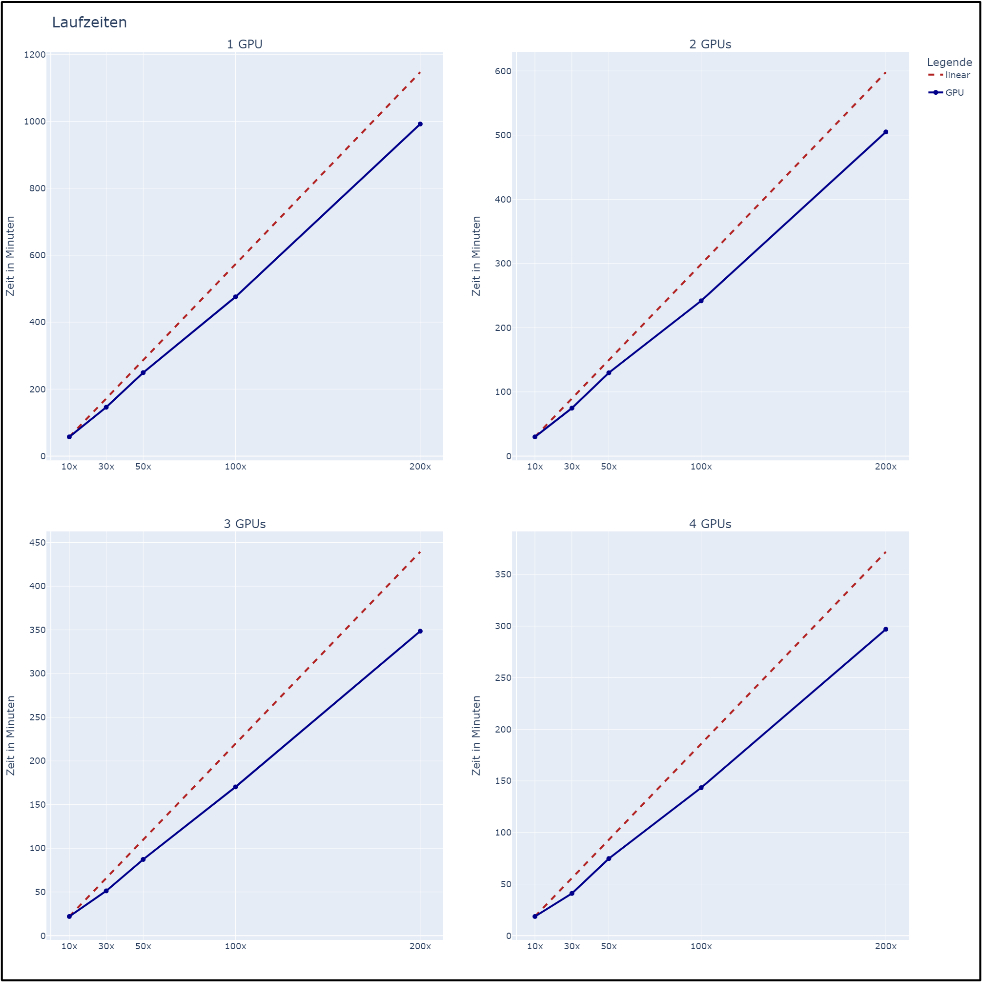
Eine Erhöhung der verwendeten GPUs führte zu einem nahezu linearen Abfall der Laufzeit (Abbildung 2). Der Überhang der verwendeten Ressourcen erhöhte sich mit jeder zusätzlichen GPU. Dadurch dauerte die Prozessierung bis zu 23 % länger als die theoretisch optimale Laufzeit, die bei vier GPUs ein Viertel der Laufzeit mit einer GPU betragen würde.
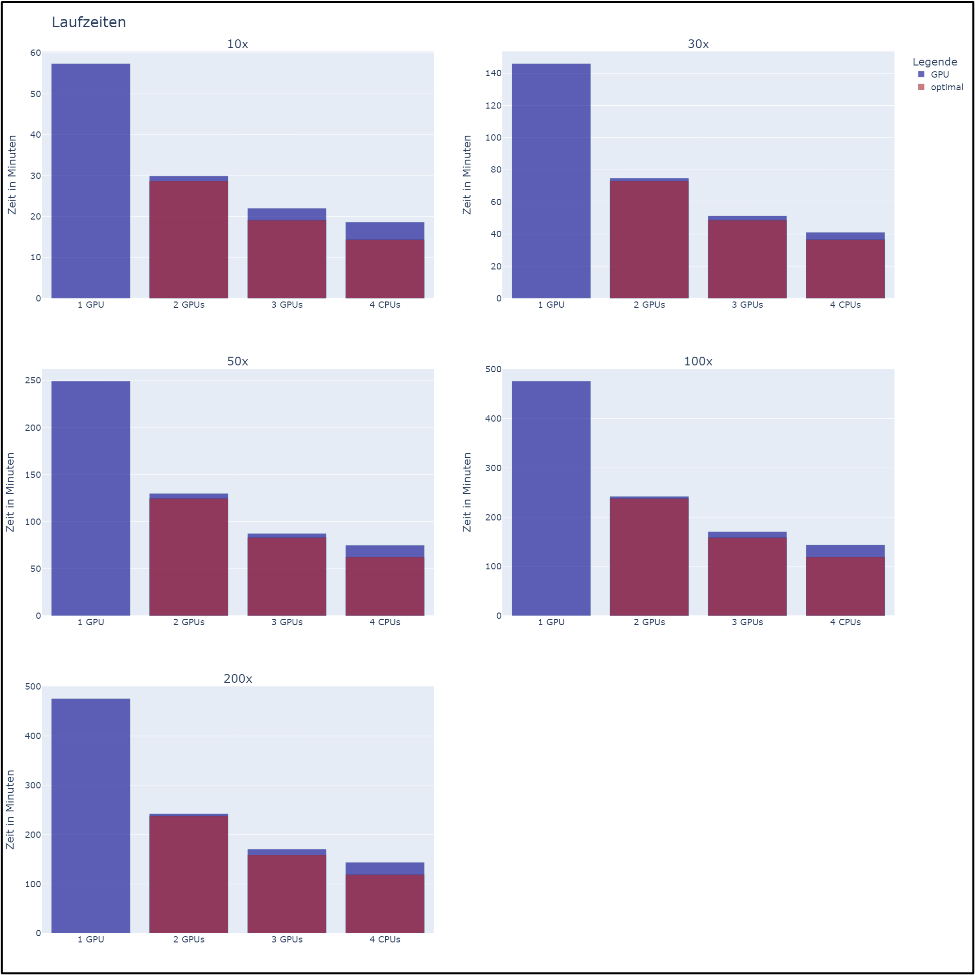
Bei einer Vergrößerung der prozessierten Daten und gleichbleibender Anzahl der GPUs verlängerte sich die Laufzeit unterproportional (Abbildung 3). Damit ist die GPU-Pipeline bei größeren Datensätzen effizienter als bei kleineren Datensätzen. Dies konnte für jede getestete Anzahl von GPUs beobachtet werden.
Vergleich der CPU- und GPU-basierten Pipelines
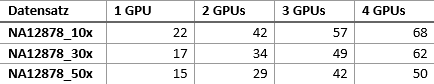
Die Verwendung der GPU-basierten Pipeline verringerte die Laufzeit um einen Faktor von 15-68 im Vergleich mit der CPU-basierten Pipeline (Tabelle 2). Die größte Beschleunigung um den Faktor 68 wurde für den kleinsten Datensatz (10x) unter Verwendung aller vier GPUs beobachtet.
Die GPU-basierte Pipeline kann in zwei Schritte aufgeteilt werden: fq2bam und haplotypecaller. Die Laufzeit des zweiten Schrittes, haplotypecaller, wurde durch die GPU-basierte Pipeline deutlich mehr reduziert als die Laufzeit des ersten Schritts (Abbildung 4).
Diskussion
Die drastische Reduzierung der Laufzeit in der sekundären Analyse von Short-Read-Sequenzdaten ermöglicht neue Anwendungsbereiche dieser Technologie. Genetische Diagnosen basierend auf Short-Read-Whole-Genome-Sequenzing sind nun eine realistische Option in der Gesundheitsversorgung, insbesondere in der Intensivmedizin. Dies setzt allerdings auch eine sehr schnelle Prozessierung der Proben in der primären Analyse im Labor voraus. Die Probenentnahme, Vorbereitung und Sequenzierung müssen entsprechend effizient ablaufen. Des Weiteren ist mit einer schnellen sekundären Analyse die Harmonisierung von Datenbeständen möglich. Um die genetische Variation einer großen Anzahl von Patienten verlässlich vergleichen zu können, müssen die zugrundeliegenden Datensätze analog prozessiert worden sein. Insbesondere die Verwendung derselben Referenz beim Alignment, aber auch dieselbe Version der verwendeten Tools ist hier wichtig. Dies ist bei Datensätzen, die zu unterschiedlichen Zeitpunkten prozessiert wurden, in der Regel nicht gegeben. Mit einer GPU-basierten Pipeline können große Datenbestände in vergleichbar kurzer Zeit erneut prozessiert werden und so zu einer vergleichenden Analyse bereitgestellt werden.
Die effiziente Nutzung von Rechenleistung bei der Verwendung von GPUs kann darüber hinaus die Kosten der sekundären Analyse weiter senken.
Grundsätzlich können CPU-basierte Pipelines stark an das verwendete System angepasst und optimiert werden. Die zu erwartende Steigerung hält sich jedoch in Grenzen und in der Regel ist der Aufwand größer als die gewonnene Leistungssteigerung. Die hier beobachteten Zeiten für das Prozessieren eines Whole-Genome-Datensatzes mit einer CPU-basierten Pipeline sind vergleichbar mit den in der Literatur beschriebenen Laufzeiten[3].
Im September 2022 wurde NVIDIA Clara Parabricks in Version 4.0 veröffentlicht. Neben einigen Updates der enthaltenen Tools entfallen seit dieser Version die Lizenzkosten und das Framework ist kostenfrei verfügbar. Die in diesem PoC verwendeten Tools sind weiterhin enthalten.
Quellen und Anhang
[2] Eberle, Michael A. et al. “A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree.” Genome research vol. 27,1 (2017): 157-164. doi:10.1101/gr.210500.116
[3] Zhao, Sen et al. “Accuracy and efficiency of germline variant calling pipelines for human genome data.” Scientific reports vol. 10,1 20222. 19 Nov. 2020, doi:10.1038/s41598-020-77218-4